โรค Creutzfeldt-Jakob (CJD): สาเหตุอาการการวินิจฉัยและการรักษา
มีจำนวนของโรคในโลกที่มีเปอร์เซ็นต์ของคนที่ได้รับผลกระทบมีขนาดเล็กมาก เหล่านี้เรียกว่าโรคที่หายาก หนึ่งในโรคที่หายากเหล่านี้คือ โรค Creutzfeldt-Jakob (CJD) ซึ่งจะกล่าวถึงตลอดบทความนี้
ในโรค Creutzfeldt-Jakob (CJD) ความผิดปกติของโปรตีนทำให้สมองมีความเสียหายที่ก้าวหน้าซึ่งส่งผลให้การทำงานและการเคลื่อนไหวของสมองลดลงอย่างรวดเร็ว ถึงคนโคม่าและความตาย
โรค Creutzfeld-Jakob คืออะไร?
โรค Creutzfeldt-Jakob (CJD) เป็นโรคสมองที่ไม่ค่อยพบซึ่งเป็นสาเหตุของความเสื่อมและคงที่ตลอดไป ถือว่าเป็นโรคที่หายากมากเนื่องจากมีผลต่อประมาณหนึ่งในล้านคน
CJD มักจะปรากฏในขั้นตอนขั้นสูงของชีวิตและมีลักษณะการพัฒนาอย่างรวดเร็ว . อาการแรกของมันมักจะปรากฏที่อายุ 60 ปีและ 90% ของผู้ป่วยเสียชีวิตหนึ่งปีหลังจากได้รับการวินิจฉัย
อาการแรกนี้คือ:
- ความล้มเหลวของหน่วยความจำ
- การเปลี่ยนแปลงพฤติกรรม
- ขาดการประสานงาน
- รบกวนภาพ
เมื่อโรคเกิดขึ้นการเสื่อมสภาพทางจิตใจจะกลายเป็นสิ่งที่มีความสำคัญมากและอาจทำให้ตาบอดการเคลื่อนไหวไม่ได้ตั้งใจความอ่อนแอในส่วนปลายและโคม่า
โรค Creutzfeldt-Jakob (CJD) สอดคล้องกับกลุ่มของโรคที่เรียกว่า encephalopathies spongiform transmissible (TSE) ในโรคเหล่านี้ สมองที่ติดเชื้อมีรูหรือรูที่สามารถมองเห็นได้เฉพาะกับกล้องจุลทรรศน์เท่านั้น ; ทำให้รูปลักษณ์คล้ายกับฟองน้ำ
สาเหตุ
ทฤษฎีทางวิทยาศาสตร์หลัก ๆ ยืนยันว่าโรคนี้ไม่ได้เกิดจากเชื้อไวรัสหรือแบคทีเรีย แต่เป็นโปรตีนประเภทหนึ่งที่เรียกว่าพรีออน
โปรตีนนี้สามารถนำเสนอทั้งปกติและไม่อันตรายรวมทั้งติดเชื้อซึ่งเป็นสาเหตุของโรค และทำให้ส่วนที่เหลือของโปรตีนร่วมกันพับในลักษณะผิดปกติมีผลต่อความสามารถในการทำงาน
เมื่อโปรตีนที่ผิดปกติเหล่านี้ปรากฏขึ้นและมารวมกันพวกเขาสร้างเส้นใยที่เรียกว่าโล่ซึ่งสามารถเริ่มสะสมเป็นเวลาหลายปีก่อนที่อาการแรกของโรคจะเริ่มปรากฏขึ้น
ประเภทของโรค Creutzfeldt-Jakob
มีสามประเภทของโรค Creutzfeldt-Jakob (CJD):
1. Sporadic CJD
เป็นชนิดที่พบมากที่สุดและปรากฏขึ้นเมื่อคนยังไม่ทราบถึงปัจจัยเสี่ยงของโรค ปรากฏใน 85% ของคดี
2. กรรมพันธุ์
เกิดขึ้นระหว่าง 5 ถึง 10 เปอร์เซ็นต์ของคดี พวกเขาเป็นคนที่มีประวัติครอบครัวเป็นโรคหรือมีการทดสอบการกลายพันธุ์ผิดปกติทางพันธุกรรมที่เกี่ยวข้อง
3. ได้มา
ไม่มีหลักฐานว่า CJD สามารถติดต่อได้โดยการติดต่อแบบสบาย ๆ กับผู้ป่วย แต่จะส่งผ่านการสัมผัสกับเนื้อเยื่อสมองหรือระบบประสาท มันเกิดขึ้นในน้อยกว่า 1% ของกรณี
อาการและพัฒนาการของโรคนี้
เริ่มแรกโรค Creutzfeldt-Jakob (CJD) ปรากฏในรูปของภาวะสมองเสื่อมกับการเปลี่ยนแปลงในบุคลิกภาพการด้อยค่าของหน่วยความจำความคิดและการฟ้องร้อง ; และในรูปแบบของปัญหาการประสานงานของกล้ามเนื้อ
เมื่อความเจ็บป่วยเกิดขึ้นการเสื่อมสภาพทางจิตใจจะทวีความรุนแรงมากขึ้น ผู้ป่วยเริ่มหดตัวกล้ามเนื้อหรือกล้ามเนื้อ myoclonus สูญเสียการควบคุมกระเพาะปัสสาวะและอาจถึงตาบอด
สุดท้ายคนสูญเสียความสามารถในการเคลื่อนย้ายและพูด; จนกระทั่งอาการโคม่าเกิดขึ้นในที่สุด ในขั้นตอนสุดท้ายนี้การติดเชื้ออื่น ๆ ที่เกิดขึ้นสามารถนำผู้ป่วยไปสู่ความตายได้
แม้ว่าอาการของ CJD อาจมีลักษณะคล้ายกับโรคความผิดปรกติของระบบประสาทเช่นโรคอัลไซเมอร์หรือฮันติงตัน แต่ CJD ก็ทำให้ความสามารถของบุคคลลดลงและมีการเปลี่ยนแปลงของเนื้อเยื่อสมองที่ไม่ซ้ำกัน พวกเขาสามารถสังเกตได้หลังจากการชันสูตรพลิกศพ
การวินิจฉัยโรค
สำหรับตอนนี้ไม่มีการทดสอบวินิจฉัยอย่างแน่ชัดสำหรับโรค Creutzfeldt-Jakob ดังนั้นการตรวจสอบจึงกลายเป็นเรื่องที่ซับซ้อนจริงๆ
ขั้นตอนแรกในการวินิจฉัยที่มีประสิทธิภาพคือการกำจัดรูปแบบอื่น ๆ ที่สามารถรักษาได้ของภาวะสมองเสื่อม สำหรับเรื่องนี้จำเป็นต้องทำการตรวจระบบประสาทที่สมบูรณ์ การตรวจอื่น ๆ ที่ใช้ในการวิเคราะห์ CJD คือการดึงกระดูกสันหลังและ electroencephalogram (EEG)
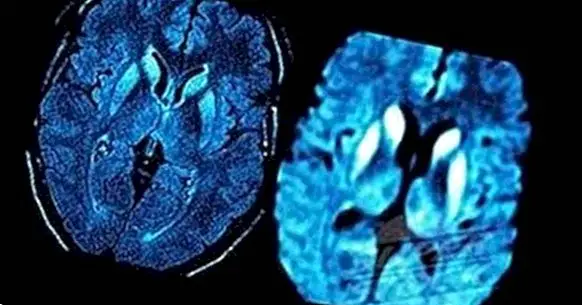
นอกจากนี้การตรวจเอกซเรย์คอมพิวเตอร์ (CT) หรือการถ่ายภาพด้วยคลื่นแม่เหล็กไฟฟ้า (MRI) ของสมองจะเป็นประโยชน์ในการวินิจฉัยว่าอาการเหล่านี้เกิดจากปัญหาอื่น ๆ เช่นเนื้องอกในสมองและระบุรูปแบบทั่วไปในการเสื่อมสภาพของสมองใน CJD
น่าเสียดายที่วิธีเดียวที่จะยืนยัน CJD คือการตรวจชิ้นเนื้อหรือการผ่าศพเนื่องจากเป็นอันตรายขั้นตอนแรกนี้จะไม่ดำเนินการเว้นแต่จะมีความจำเป็นที่จะต้องใช้หลักเกณฑ์ใด ๆ ที่สามารถรักษาได้ นอกจากนี้ ความเสี่ยงของการติดเชื้อจากขั้นตอนเหล่านี้ทำให้พวกเขามีความซับซ้อนมากขึ้นในการดำเนินการ .
การรักษาและการพยากรณ์โรค
เช่นเดียวกับไม่มีการทดสอบวินิจฉัยโรคนี้ไม่มีการรักษาที่สามารถรักษาหรือควบคุมได้
ปัจจุบันผู้ป่วยที่ได้รับ CJD ได้รับการรักษาแบบประคับประคองโดยมีวัตถุประสงค์หลักในการบรรเทาอาการของผู้ป่วยและทำให้ผู้ป่วยได้รับคุณภาพชีวิตที่ดีที่สุด สำหรับกรณีเหล่านี้การใช้ยาเสพติดยาเสพติดยาเสพติด clonazepam และโซเดียม valproate สามารถช่วยลดอาการปวดและบรรเทา myoclonus สำหรับการพยากรณ์โรคแนวโน้มของคนป่วยของ CJD ค่อนข้างท้อใจ หลังจากหกเดือนหรือน้อยกว่าหลังจากเริ่มมีอาการผู้ป่วยไม่สามารถดูแลตัวเองได้
โดยทั่วไป ความผิดปกติกลายเป็นอันตรายถึงชีวิตได้ในระยะเวลาสั้น ๆ ประมาณแปดเดือน ; แม้ว่าบางส่วนของประชากรจะมีชีวิตรอดได้หนึ่งหรือสองปีก็ตาม
สาเหตุส่วนใหญ่ของการเสียชีวิตใน CJD คือการติดเชื้อและความผิดปกติของหัวใจหรือทางเดินหายใจ
วิธีการถ่ายทอดและวิธีการหลีกเลี่ยง
ความเสี่ยงต่อการแพร่เชื้อ CJD ต่ำมาก เป็นหมอที่ทำงานกับสมองหรือเนื้อเยื่อประสาทผู้ที่สัมผัสกับมันมากที่สุด
โรคนี้ไม่สามารถส่งผ่านทางอากาศหรือผ่านการติดต่อกับคนที่ทนทุกข์ทรมาน อย่างไรก็ตาม การติดต่อทางตรงหรือทางอ้อมกับเนื้อเยื่อสมองและไขสันหลังปิดเป็นความเสี่ยง .
เพื่อหลีกเลี่ยงความเสี่ยงต่อการติดเชื้อในผู้ป่วยคนที่สงสัยหรือได้รับการวินิจฉัยว่าเป็น CJD อยู่แล้วไม่ควรบริจาคโลหิตเนื้อเยื่อหรืออวัยวะต่างๆ
สำหรับคนที่ดูแลผู้ป่วยเหล่านี้ผู้เชี่ยวชาญด้านสุขภาพและแม้แต่ผู้เชี่ยวชาญด้านการฝังศพต้องนำข้อควรระวังต่างๆมาใช้ในการปฏิบัติงาน บางส่วนของเหล่านี้คือ:
- ล้างมือและผิวหนังที่สัมผัส
- ตัดผ้าหรือรอยถลอกด้วยผ้าพันแผลกันน้ำ
- สวมถุงมือผ่าตัดเมื่อจัดการเนื้อเยื่อและของเหลวของผู้ป่วย
- ใช้อุปกรณ์ป้องกันใบหน้าและผ้าปูที่นอนหรือผ้าอ้อมอื่น ๆ
- ทำความสะอาดเครื่องมือที่ใช้ในการแทรกแซงหรือสัมผัสกับผู้ป่วย